New Microscopic Properties of Magnetic Reconnection Derived by Cluster
19 May 2006
Magnetic reconnection is considered to be the most efficient mechanism for solar material to penetrate the Earth's magnetic shield and to convert magnetic energy to particle energy (generating plasma jets and plasma heating). However, our understanding of this fundamental physical process is far from being complete. High resolution data by the Cluster mission reveal new microscopic properties at the heart of the reconnection process, in a region called the electron diffusion region. Published 17 December 2005, in Geophysical Research Letters, these multipoint measurements enable, in particular, a direct estimation of the spatial thickness of this central region.
|
Image 1. This picture is a false-color image of an X-17 solar flare on 28 October 2003, made with SOHO's Extreme ultraviolet Imaging Telescope (EIT). X-class flares are the most powerful category, capable of releasing the energy of a billion one-megaton nuclear bombs. The flare is the large, white area in the bottom half of the solar disk. The horizontal lines resulted when the flare's intense light saturated the EIT instrument (Credit: ESA and NASA) Animation 1. Sequence of EIT images, showing the X-17 class solar flare of 28 October 2003. The ejected material was sent hurtling towards Earth at a speed of over 2000 kilometres per second. |
|


On 28 October 2003, the ESA/NASA SOHO spacecraft captured one of the most powerful solar flares ever recorded in recent years [Image 1 and Animation 1]. This tremendous solar explosion was 50 billion times more powerful than an atomic bomb. It triggered the release of one billion tonnes of hot plasma towards the Earth and affected many satellites.
 |
|
Animation 2. Reconnecting magnetic field lines |
 |
|
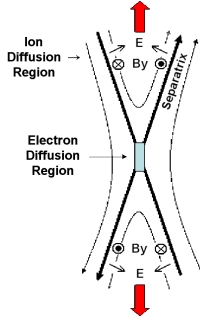
Image 2. Electron diffusion region sketch (courtesy of Prof. Forrest Mozer, UC Berkeley, USA) |
Magnetic reconnection does not only occur at the Sun. It is a universal phenomenon observed in astrophysical plasmas (for example at neutron stars, accretion disks), planetary magnetospheres (such as around Saturn, Jupiter, Earth) and in the solar wind. This fundamental process of plasma physics has also important relevance to controlled nuclear fusion, a nuclear waste free technology (under development) for tomorrow's electricity-producing power plants. It is indeed a mechanism preventing magnetic confinement of the fusion fuel. However, our understanding of reconnection is far from being complete. By studying it in space, we have access to an open laboratory enabling us to derive some of its fundamental properties, later on applicable to more confined laboratory plasmas.
Magnetic reconnection is a multi-scale physical process. On one hand, it is a macroscopic process that can affect large volumes of plasma in space (for example solar wind regions larger than 2.5 million kilometres wide [Phan et al., 2006]). At the same time, most of the magnetic field topology changes occur within small localised regions (less than 100 km). One of these key regions is located at the heart of the reconnection process: the electron diffusion region [Image 2].
 |
|
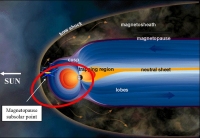
Image 3. Sketch of the Earth's magnetosphere, with the Cluster orbit indicated by the red ellipse (credit: ESA) |
"Experimentally, direct encounters with electron diffusion regions have been rare and details of these regions have not been well resolved", wrote Forrest Mozer, Professor Emeritus at the Space Sciences Laboratory, UC Berkeley (USA) and co-investigator of the Electric Field and Wave (EFW) experiment [Gustafsson et al., 1997] onboard each of the four Cluster spacecraft. On 25 January 2005, the Cluster space fleet was flying near the subsolar magnetopause [Image 3], the external boundary layer of the magnetosphere, bounded by two separatrices: the magnetosheath separatrix and the magnetospheric separatrix. The magnetopause can contain several electron diffusion regions [Mozer, 2005b].
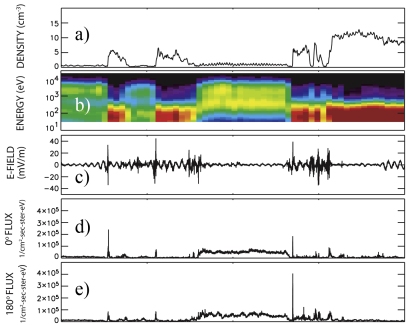
In a paper published 17 December 2005 in Geophysical Research Letters, Forrest Mozer and co-workers demonstrate the presence of 19 electron diffusion regions, all located at the magnetospheric separatrix, by analyzing one hour of data collected on 25 January 2005. These regions were found by searching for areas with electron beam acceleration and high electromagnetic energy, among other factors. Their analysis showed for the first time that these regions directly convert electromagnetic energy to accelerated electrons. In particular, thanks to high time resolution measurements, the first direct observation of electron beams accelerated in the electron diffusion region is demonstrated [Image 4]. The time resolution of one component of the raw electric field measured by EFW is 2.2 milliseconds (panel c). The fluxes of the electrons moving parallel (panel d) and anti-parallel (panel e) to the magnetic field were measured every 8 milliseconds by the Electron Drift Instrument [Paschmann et al., 2001].
 |
|
Image 4. Plasma and field parameters measured on Cluster spacecraft 2 during a four minute interval: plasma density inferred from the EFW spacecraft potential measurements (panel a); electron energy flux integrated over solid angle measured by the PEACE instrument (panel b); one component of the raw electric field measured by the EFW instrument (panel c); fluxes of the electrons moving parallel (panel d) and anti-parallel (panel e) to the magnetic field, measured by the EDI instrument (Excerpt of Figure 1 of Mozer et al., 2005, courtesy of Prof. Mozer, UC Berkeley, USA). |
Making use of the unique multi-point capability of Cluster, the authors were also able to estimate the thicknesses of these regions quantitatively (~ 5 km). Further analysis revealed that these regions were boundaries between open and closed magnetic field geometries that contained different plasma flows and magnetic field lines.
"Understanding the microphysics of magnetic reconnection is essential to understand its macroscopic consequences. However, the 3-dimensional nature of this physical process implies that some of its fundamental microscopic properties are only accessible through closely located multi-point measurements. Cluster, the first magnetospheric mission composed of 4 spacecraft flying in close formation, is revolutionising our understanding of magnetic reconnection and will continue to do so in the years to come. It is also paving the way to future formation flying missions like MMS (Magnetospheric MultiScale mission) or Cross-Scale", says Philippe Escoubet, Cluster and Double Star project scientist of the European Space Agency.
Mozer, F. S., S. D. Bale, J. P. McFadden, and R. B. Torbert (2005), New features of electron diffusion regions observed at subsolar magnetic field reconnection sites, Geophys. Res. Lett., 32, L24102, doi:10.1029/2005GL024092.
Related Articles
Phan, T. D. , J. T. Gosling, M. S. Davis, R. M. Skoug, M. Øieroset, R. P. Lin, R. P. Lepping, D. J. McComas, C. W. Smith, H. Rème, A. Balogh, A magnetic reconnection X-line extending more than 390 Earth radii in the solar wind, Nature 439, 175-178, 2006Paschmann, G., J.M. Quinn, R.B. Torbert, H. Vaith, C.E. McIlwain, G. Haerendel, O.H. Bauer, T. Bauer, W. Baumjohann, W. Filliius, M. Förster, S. Frey, S.S. Kerr, C.A. Kletzing, P. Puhl-Quinn, and E.C. Whipple, The electron drift instrument on Cluster: overview of first results, Ann. Geophysicae, 19, 1273, 2001
Gustafsson et al., The electric field and wave experiment for the Cluster mission, Space Sci. Rev., 79, 137, 1997.
Vaivads, A., Retinò, A. and André M., Microphysics of magnetic reconnection, Space Science Reviews, in press, 2006
Mozer, F. S. (2005b), Criteria for and statistics of electron diffusion regions associated with subsolar magnetic field reconnection, J. Geophys. Res., 110, A12222, doi:10.1029/2005JA011258
Magnetic reconnection region larger than 2.5 million kilometres found in the solar wind
http://sci.esa.int/jump.cfm?oid=38574
Direct observation of 3D magnetic reconnection
http://sci.esa.int/jump.cfm?oid=36447
Contact
Forrest MozerSpace Sciences Laboratory, University of California, Berkeley, USA
Tel: +1-510-642-0549
Email: fmozer
 ssl.berkeley.edu
ssl.berkeley.edu
Web story author and co-editor
Arnaud Masson
Research and Scientific Support Department, ESA, The Netherlands
Tel: +31-71-565-5634
Email: Arnaud.Massonesa.int
Web story editor
Philippe Escoubet
Research and Scientific Support Department, ESA, The Netherlands
Tel: +31-71-565-3454
Email: Philippe.Escoubetesa.int
